Performing a Genome Scan with Binary Traits
Last updated on 2025-10-07 | Edit this page
Estimated time: 50 minutes
Overview
Questions
- How do I perform a genome scan for binary traits?
Objectives
- Convert phenotypes to binary values.
- Use logistic regression for genome scans with binary traits.
- Plot and compare genome scans for binary traits.
Binary Phenotypes
The genome scans in the previous episode were performed assuming that
the residual variation followed a normal distribution. This will often
provide reasonable results even if the residuals are not normal, but an
important special case is that of a binary trait, with values 0 and 1,
which is best treated differently. The scan1 function can
perform a genome scan with binary traits by logistic regression, using
the argument model="binary". (The default value for the
model argument is "normal"). At present, we
cannot account for kinship relationships among individuals in
this analysis.
Let’s look at the phenotypes in the cross again.
R
head(cross$pheno)
OUTPUT
log10_insulin_10wk agouti_tan tufted
Mouse3051 1.399 1 0
Mouse3551 0.369 1 1
Mouse3430 0.860 0 1
Mouse3476 0.800 1 0
Mouse3414 1.370 0 0
Mouse3145 1.783 1 0There are two binary traits called agouti_tan and
tufted which are related to coat color and shape.
The agouti_tan phenotype is 1 if a mouse
has an agouti or tan coat and 0 if a mouse has a black
coat. The founder strains have different coat colors. C57BL/6J has a
black coat.

BTBR appears to have a black coat, but this coat color is actually called “black and tan” because their bellies are tan.

Agouti mice appear to be tan or brown, but their hair is a mix of brown and black coloring. As an example, C3H/HeJ mice have agouti coats.

There is more information about mouse coat colors in the JAX Coat Color guide.
The tufted phenotype has to do with progressive hair loss in a
“tufted” pattern. Mice with a value of 1 have the tufted
phenotype and mice with a value of 0 do not.

Photo Credit: Ellis et al., J Hered, 2013
Performing a Binary Genome Scan
We perform a binary genome scan in a manner similar to mapping
continuous traits by using scan1. When we mapped insulin,
there was a hidden argument called model which told
qtl2 which mapping model to use. There are two options:
normal, the default, and binary. The
normal argument tells qtl2 to use a
normal (least squares) linear model. To map a binary trait, we
will include the model = "binary" argument to indicate that
the phenotype is a binary trait with values 0 and 1.
R
lod_agouti <- scan1(genoprobs = probs,
pheno = cross$pheno[,'agouti_tan', drop = FALSE],
addcovar = addcovar,
model = "binary")
Let’s plot the result and see if there is a peak.
R
plot_scan1(x = lod_agouti,
map = cross$pmap,
main = 'Agouti')
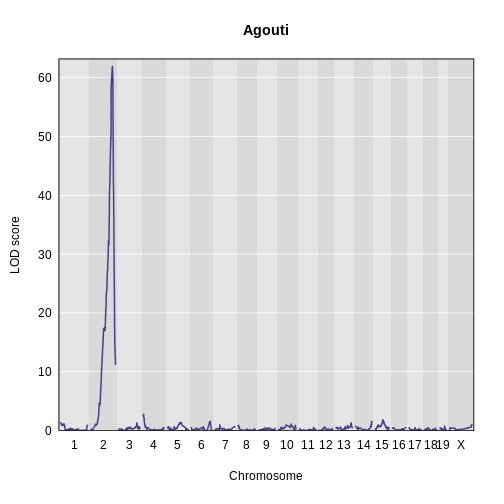
Yes! There is a big peak on chromosome 2. Let’s zoom in on chromosome 2.
R
plot_scan1(x = lod_agouti,
map = cross$pmap,
chr = "2",
main = "Agouti")
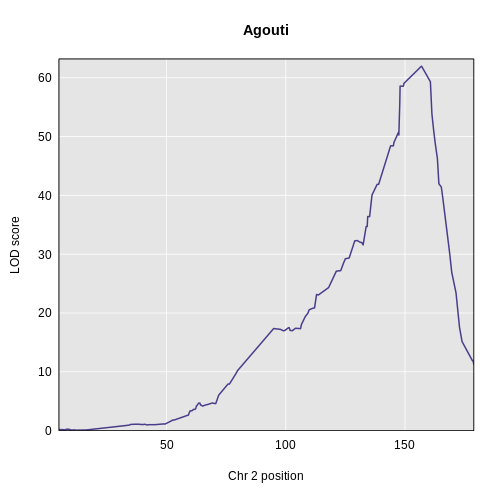
We can use find_peaks to find the position of the
highest LOD score.
R
find_peaks(scan1_output = lod_agouti,
map = cross$pmap)
OUTPUT
lodindex lodcolumn chr pos lod
1 1 agouti_tan 2 157 61.9This turns out to be a well-known coat color locus for agouti coat color which contains the nonagouti gene. Mice carrying two black alleles will have a black coat, and mice carrying one or no black alleles will have agouti coats.
Challenge 1: How many mice have black coats?
Look at the frequency of the black (0) and agouti (1) phenotypes.
What proportion of the mice are black? Can you use what you learned
about how the nonagouti locus works and the cross design to
explain the frequency of black mice?
First, get the number of black and agouti mice.
R
tbl <- table(cross$pheno[,"agouti_tan"])
tbl
OUTPUT
0 1
125 356 Then use the number of mice to calculate the proportion with each coat color.
R
tbl / sum(tbl)
OUTPUT
0 1
0.26 0.74 We can see that the black (0) mice occur about 25 % of the time. If
the B allele causes mice to have black coats when it is
recessive, and if R is the agouti allele, then, when
breeding two heterozygous (BR) mice together, we expect the
following genotypes in the progeny:
| / | B | R |
|---|---|---|
| B | BB | BR |
| R | BR | RR |
Hence, we expect mean allele frequencies and coat colors as follows:
| Allele | Frequency | Coat Color |
|---|---|---|
| BB | 0.25 | black |
| BR | 0.50 | agouti |
| RR | 0.25 | agouti |
From this, we can see that about 25% of the mice should have black coats.
Challenge 2: Map the “tufted” phenotype.
Map the tufted phenotype an determine if there are any tall peaks for this trait.
First, map the trait.
R
lod_tufted <- scan1(genoprobs = probs,
pheno = cross$pheno[,"tufted", drop = FALSE],
addcovar = addcovar,
model = "binary")
Then, plot the LOD score.
R
plot_scan1(x = lod_tufted,
map = cross$pmap,
main = "Tufted")
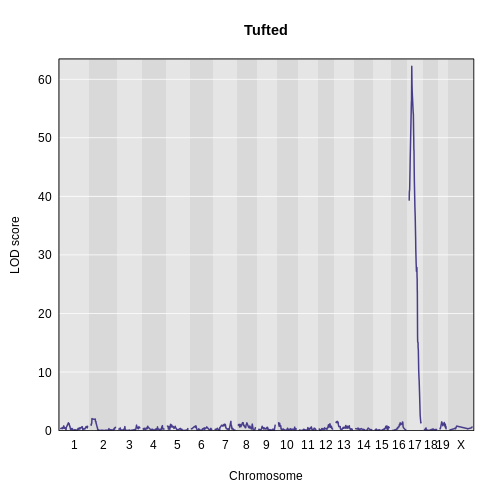
Finally, use find_peaks to get the peak LOD
location.
R
find_peaks(scan1_output = lod_tufted,
map = cross$pmap)
OUTPUT
lodindex lodcolumn chr pos lod
1 1 tufted 17 27.3 62.2There is a large peak on chromosome 17. This is a known locus associated with the Itpr3 gene near 27.3 Mb on chromosome 17.
- A genome scan for binary traits (0 and 1) requires special handling; scans for non-binary traits assume normal variation of the residuals.
- A genome scan for binary traits is performed using logistic regression.