Maximum eQTL Peaks and Nearby Genes
Last updated on 2025-09-16 | Edit this page
Estimated time: 40 minutes
Overview
Questions
- How do I select QTL peaks for each gene?
- How do I find genes near QTL peaks?
Objectives
- Learn how to filter LOD peaks to retain significant peaks.
- Understand the two levels of multiple testing in eQTL mapping studies.
- Find genes which are located under QTL peaks.
Multiple Testing in QTL Mapping
When we perform multiple hypothesis tests, as we do in QTL mapping, we must adjust for the fact that our overall false-positive rate is higher than the nominal false-positive rate for a single test. There are also two levels of multiple testing in expression QTL mapping, at the marker level and at the gene level.
When we perform a single QTL scan, we are performing a hypothesis test at each marker. We could perform a traditional multiple-testing correction such as a Benjamini-Hochberg false discovery rate (FDR). However, by performing permutations of the sample labels, scanning the genome, and retaining the maximum LOD from each permutation, we are effectively adjusting for multiple testing across the genome because we are selecting only the maximum LOD across all markers in each permutation.
Multiple testing at the gene level comes from mapping multiple genes. If we use the 0.05 significance level for all genes, our overall false-positive rate will be greater than 0.05. We must also perform a multiple testing correction for each gene. However, we have more than one peak per gene, so how should we proceed?
While genes that have multiple peaks are important and may have interesting biology, we do not currently have a method of adjusting for multiple QTL peaks for each gene. Instead, we will select the peak with the highest LOD for each gene and will then perform a multiple-testing correction.
First, we will filter the peaks to retain the peak with the highest LOD score for each gene.
R
peaks_filt <- peaks |>
group_by(lodcolumn) |>
filter(lod == max(lod))
Let’s make sure that we have only one peak per gene.
R
peaks_filt |>
ungroup() |>
dplyr::count(lodcolumn) |>
dplyr::count(n)
OUTPUT
# A tibble: 1 × 2
n nn
<int> <int>
1 1 21771This looks good! All of the genes have only one peak.
Note that only 21771 genes out of 21771 have QTL peaks.
Most multiple-testing correction methods rely upon p-values as input.
We currently have LOD scores. We will need to convert them into
genome-wide p-values using the permutations that we performed in Mapping
One eQTL. We have these in a variable called eperm. We will
estimate the p-value for each gene by getting the proportion of
permutations with a LOD greater than or equal to the LOD of each
gene.
Challenge 1: What is the minimum p-value that we can have?
We performed 1,000 permutations. What does this tell you about the minimum possible p-value that we can get?
Technically, we could get a p-value of 0 if the gene’s LOD score is above the highest LOD score in the permutations. In practice, we recognize that the p-value isn’t zero, but some number less than one over the number of permutations (1 / 1000) that we performed.
Let’s estimate the p-values by calculating the proportion of permutations with LOD scores greater than or equal to each gene’s LOD. We will also adjust p-values which are zero to be 0.001 to be conservative.
R
peaks_filt <- peaks_filt |>
group_by(lodcolumn) |>
mutate(pvalue = mean(lod <= eperm[,1]),
pvalue = if_else(pvalue < 0.001, 0.001, pvalue))
Now we can apply a procedure called q-value to estimate the false discovery rate (FDR) for each gene.
R
peaks_filt <- peaks_filt |>
ungroup() |>
mutate(qvalue = qvalue(peaks_filt$pvalue)$qvalue)
Now we can plot the nominal p-values versus the q-values.
R
peaks_filt |>
ggplot(aes(pvalue, qvalue)) +
geom_line(linewidth = 1.25) +
geom_abline(aes(intercept = 0, slope = 1), color = 'red', linewidth = 1.25) +
labs(title = "p-values versus q-values") +
theme(text = element_text(size = 20))
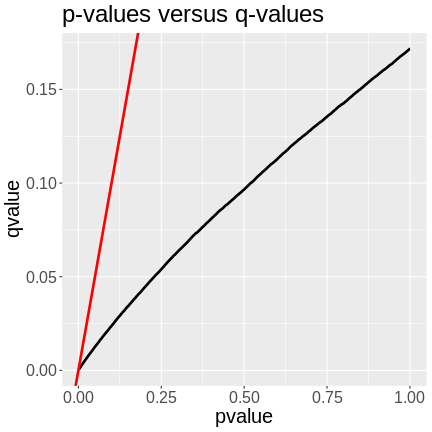
In the plot above, we can see that the q-values are lower than the p-values. The p-value versus q-value line is shown in black and the x = y line in red. The q-values represent the FDR of each LOD peak and all of the genes with lower q-values. A p-value of 0.25 has a q-value of ~0.06, meaning that the FDR of LOD peaks with p-values of 0.25 or less have an FDR or 0.06.
We can filter the list of peaks to include ones with q-values less than or equal to 0.05.
R
peaks_filt <- peaks_filt |>
filter(qvalue <= 0.05)
Let’s see how many genes we have retained.
R
nrow(peaks_filt)
OUTPUT
[1] 17089We still have almost 17,000 genes with an FDR of 5% or less.
Next, let’s look at what the range of LOD scores is.
R
range(peaks_filt$lod)
OUTPUT
[1] 6.646723 189.043226The lowest LOD score is 6.6467234, which is lower than the permutation threshold of 7.4745761.
Finding Genes under QTL Peaks
We can use the gene annotation data to find genes under QTL peaks. Let’s get the peak with the highest LOD score.
R
high_peak <- slice_max(peaks_filt, order_by = lod, n = 1)
high_peak
OUTPUT
# A tibble: 1 × 9
lodindex lodcolumn chr pos lod ci_lo ci_hi pvalue qvalue
<int> <chr> <fct> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 2230 ENSMUSG00000020268 11 54.8 189. 54.8 55.1 0.001 0.000311DMG: This is temporary. Remove later.
R
high_peak$ci_lo = 106
high_peak$ci_hi = 107
This peak has a LOD of 189.043226! Next, we will get the genes which lie within the confidence interval from the gene annotation.
Let’s remind ourselves what the gene annotation contains.
R
head(annot)
OUTPUT
gene_id symbol chr start end strand
ENSMUSG00000000001 ENSMUSG00000000001 Gnai3 3 108.10728 108.14615 -1
ENSMUSG00000000028 ENSMUSG00000000028 Cdc45 16 18.78045 18.81199 -1
ENSMUSG00000000037 ENSMUSG00000000037 Scml2 X 161.11719 161.25821 1
ENSMUSG00000000049 ENSMUSG00000000049 Apoh 11 108.34335 108.41440 1
ENSMUSG00000000056 ENSMUSG00000000056 Narf 11 121.23725 121.25586 1
ENSMUSG00000000058 ENSMUSG00000000058 Cav2 6 17.28119 17.28911 1
middle nearest.marker.id biotype module
ENSMUSG00000000001 108.12671 3_108090236 protein_coding darkgreen
ENSMUSG00000000028 18.79622 16_18817262 protein_coding grey
ENSMUSG00000000037 161.18770 X_161182677 protein_coding grey
ENSMUSG00000000049 108.37887 11_108369225 protein_coding greenyellow
ENSMUSG00000000056 121.24655 11_121200487 protein_coding brown
ENSMUSG00000000058 17.28515 6_17288298 protein_coding brown
hotspot
ENSMUSG00000000001 <NA>
ENSMUSG00000000028 <NA>
ENSMUSG00000000037 <NA>
ENSMUSG00000000049 <NA>
ENSMUSG00000000056 <NA>
ENSMUSG00000000058 <NA>We can use the start, middle, or end columns to get the gene positions. In this case, we will use the middle.
R
annot_filt <- annot |>
filter(chr == high_peak$chr &
middle >= high_peak$ci_lo &
middle <= high_peak$ci_hi)
There are 35 genes within the QTL support interval. This is a large number and would require more research to find candidate genes.
Let’s see where the gene being mapped is located. Note that the
Ensembl ID of the gene is in high_peak.
R
filter(annot, gene_id == high_peak$lodcolumn)
OUTPUT
gene_id symbol chr start end strand
ENSMUSG00000020268 ENSMUSG00000020268 Lyrm7 11 54.82687 54.86092 -1
middle nearest.marker.id biotype module hotspot
ENSMUSG00000020268 54.84389 11_54852726 protein_coding grey <NA>Challenge 2: Where is the gene?
Look at the gene location above and compare it with its corresponding
QTL peak in high_peak. Is there any relationship between
the two genomic positions?
Print out the QTL peak position.
R
high_peak
OUTPUT
# A tibble: 1 × 9
lodindex lodcolumn chr pos lod ci_lo ci_hi pvalue qvalue
<int> <chr> <fct> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 2230 ENSMUSG00000020268 11 54.8 189. 106 107 0.001 0.000311The QTL is located on chromosome 11 at 54.795542 Mb. The gene is located on chromosome 11 at Mb. These genomic positions are nearly identical.
- There can be more than one significant QTL peak for each gene.
- We sometimes focus on the largest peak for each gene.
- A multiple-testing correction should be applied to all peaks.
- Sometimes a gene s co-located with its QTL peak.