Mapping A Single Gene Expression Trait
Overview
Teaching: 30 min
Exercises: 30 minQuestions
How do I map one gene expression trait?
Objectives
QTL mapping of an expression data set
In this tutorial, we are going to use the gene expression data as our phenotype to map QTLs. That is, we are looking to find any QTLs (cis or trans) that are responsible for variation in gene expression across samples.
We are using the same directory as our previous tutorial, which you can check by typing getwd() in the Console. If you are not in the correct directory, type in setwd("code") in the console or Session -> Set Working Directory -> Choose Directory in the RStudio menu to set your working directory to the main directory.
If you have started a new R session, you will need to load the libraries again. If you haven’t, the libraries listed below are already loaded.
Load Libraries
library(tidyverse)
library(knitr)
library(broom)
library(qtl2)
Load Data
In this lesson, we are loading in gene expression data for 21,771 genes: attie_DO500_expr.datasets.RData that includes normalised and raw gene expression data. dataset.islet.rnaseq is the dataset you can download directly from Dryad. Again, if you are using the same R session, you will not need to the load the mapping, phenotypes and genotype probabilities data, again.
# expression data
load("../data/attie_DO500_expr.datasets.RData")
# data from paper
load("../data/dataset.islet.rnaseq.RData")
# phenotypes
load("../data/attie_DO500_clinical.phenotypes.RData")
# mapping data
load("../data/attie_DO500_mapping.data.RData")
# genotype probabilities
probs = readRDS("../data/attie_DO500_genoprobs_v5.rds")
Expression Data
Raw gene expression counts are in the counts data object. These counts have been normalised and saved in the norm data object. More information is about normalisation is here.
To view the contents of either one of these data , click the table on the right hand side either norm or counts in the Environment tab. If you type in names(counts), you will see the names all start with ENSMUSG. These are Ensembl IDs. If we want to see which gene these IDs correspond to, type in dataset.islet.rnaseq$annots, which gives information about each gene, including ensemble id, gene symbol as well as start & stop location of the gene and chromsome on which the gene lies.
Because we are working with the insulin tAUC phenotype, let’s map the expression counts for Hnf1b which is known to influence this phenotype is these data. First, we need to find the Ensembl ID for this gene:
dataset.islet.rnaseq$annots[dataset.islet.rnaseq$annots$symbol == "Hnf1b",]
gene_id symbol chr start end strand
ENSMUSG00000020679 ENSMUSG00000020679 Hnf1b 11 83.85006 83.90592 1
middle nearest.marker.id biotype module
ENSMUSG00000020679 83.87799 11_84097611 protein_coding midnightblue
hotspot
ENSMUSG00000020679 <NA>
We can see that the ensembl ID of Hnf1b is ENSMUSG00000020679. If we check the distribution for Hnf1b expression data between the raw and normalised data, we can see there distribution has been corrected. Here is the distribution of the raw counts:
hist(counts$ENSMUSG00000020679, main = "Hnf1b")
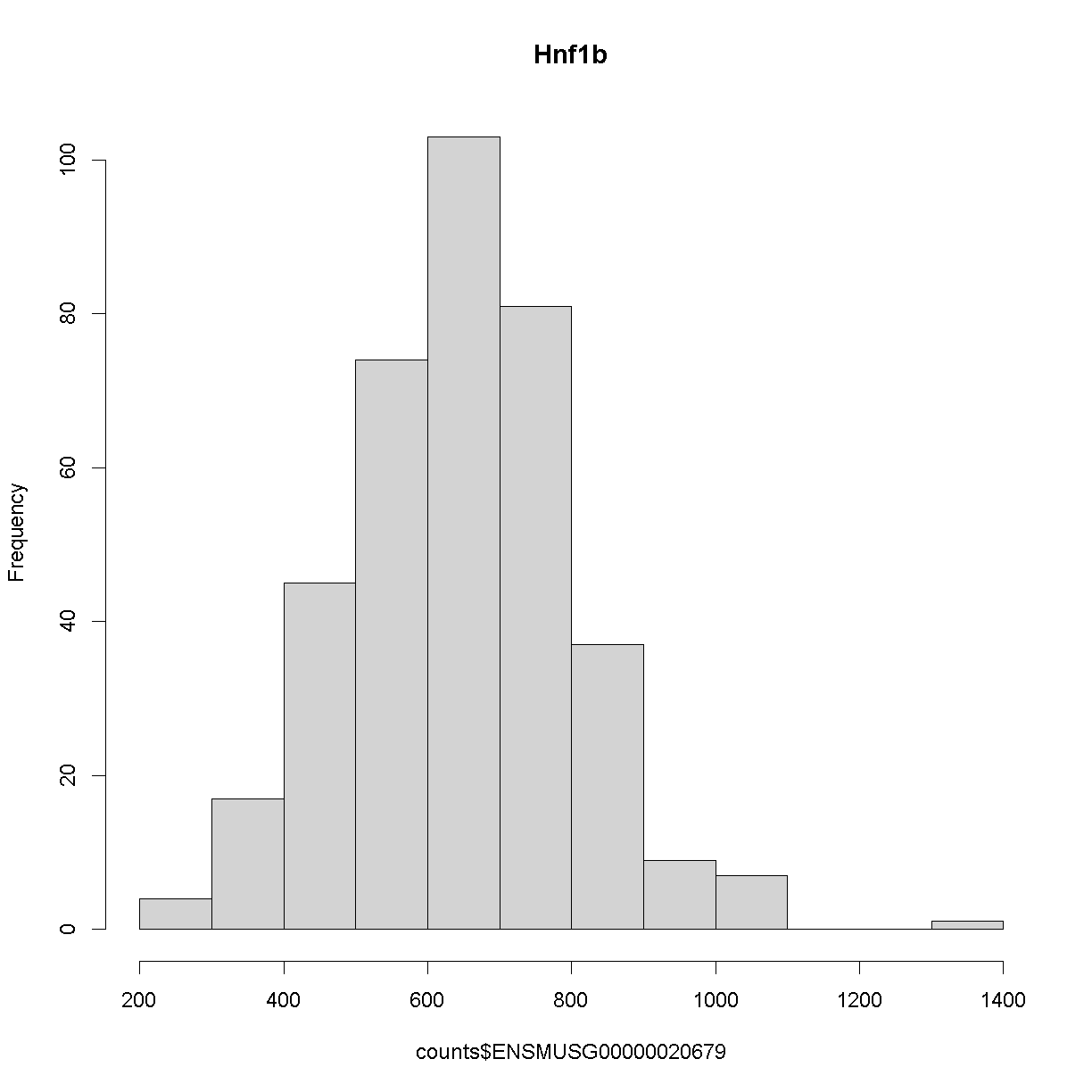
and here is the distribution of the normalised counts:
hist(norm$ENSMUSG00000020679, main = "Hnf1b")
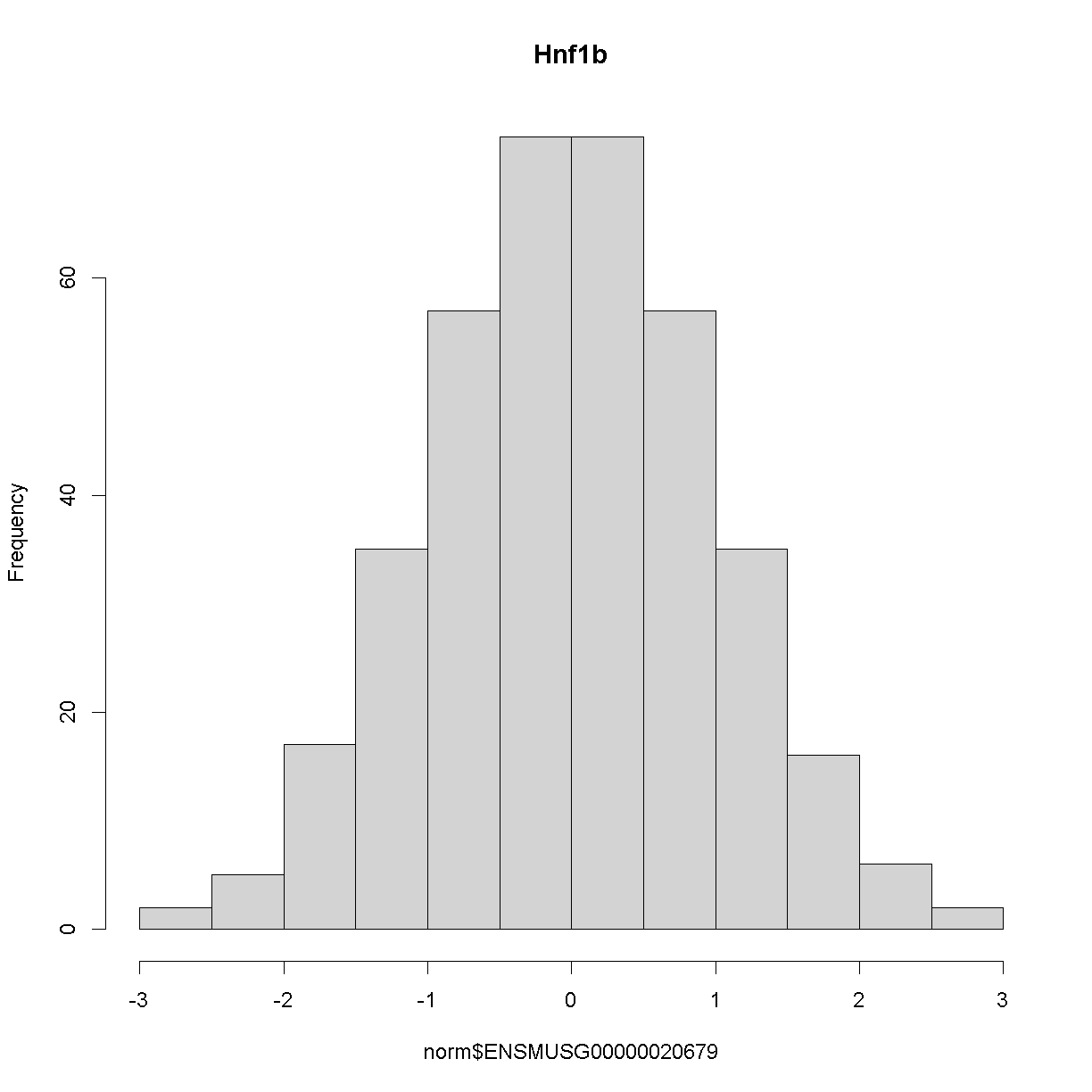
The histogram indicates that distribution of these counts are normalised.
The Marker Map
We are using the same marker map as in the previous lesson
Genotype probabilities
We have explored this earlier in th previous lesson. But, as a reminder, we have already calculated genotype probabilities which we loaded above called probs. This contains the 8 state genotype probabilities using the 69k grid map of the same 500 DO mice that also have clinical phenotypes.
Kinship Matrix
We have explored the kinship matrix in the previous lesson. It has already been calculated and loaded in above.
Covariates
Now let’s add the necessary covariates. For Hnf1b expression data, let’s see which covariates are significant.
###merging covariate data and expression data to test for sex, wave and diet_days.
cov.counts <- merge(covar, norm, by=c("row.names"), sort=F)
#testing covairates on expression data
tmp = cov.counts %>%
dplyr::select(mouse, sex, DOwave, diet_days, ENSMUSG00000020679) %>%
gather(expression, value, -mouse, -sex, -DOwave, -diet_days) %>%
group_by(expression) %>%
nest()
mod_fxn = function(df) {
lm(value ~ sex + DOwave + diet_days, data = df)
}
tmp = tmp %>%
mutate(model = map(data, mod_fxn)) %>%
mutate(summ = map(model, tidy)) %>%
unnest(summ)
# kable(tmp, caption = "Effects of Sex, Wave & Diet Days on Expression")
tmp
# A tibble: 4 x 8
# Groups: expression [1]
expression data model term estimate std.e~1 stati~2 p.value
<chr> <list> <list> <chr> <dbl> <dbl> <dbl> <dbl>
1 ENSMUSG00000020679 <tibble> <lm> (Interce~ -1.70 0.506 -3.36 8.72e- 4
2 ENSMUSG00000020679 <tibble> <lm> sexM -0.199 0.0824 -2.42 1.60e- 2
3 ENSMUSG00000020679 <tibble> <lm> DOwave 0.510 0.0370 13.8 3.73e-35
4 ENSMUSG00000020679 <tibble> <lm> diet_days 0.00403 0.00386 1.04 2.97e- 1
# ... with abbreviated variable names 1: std.error, 2: statistic
tmp %>%
filter(term != "(Intercept)") %>%
mutate(neg.log.p = -log10(p.value)) %>%
ggplot(aes(term, neg.log.p)) +
geom_point() +
facet_wrap(~expression) +
labs(title = "Significance of Sex, Wave & Diet Days on Expression") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5)) +
rm(tmp)
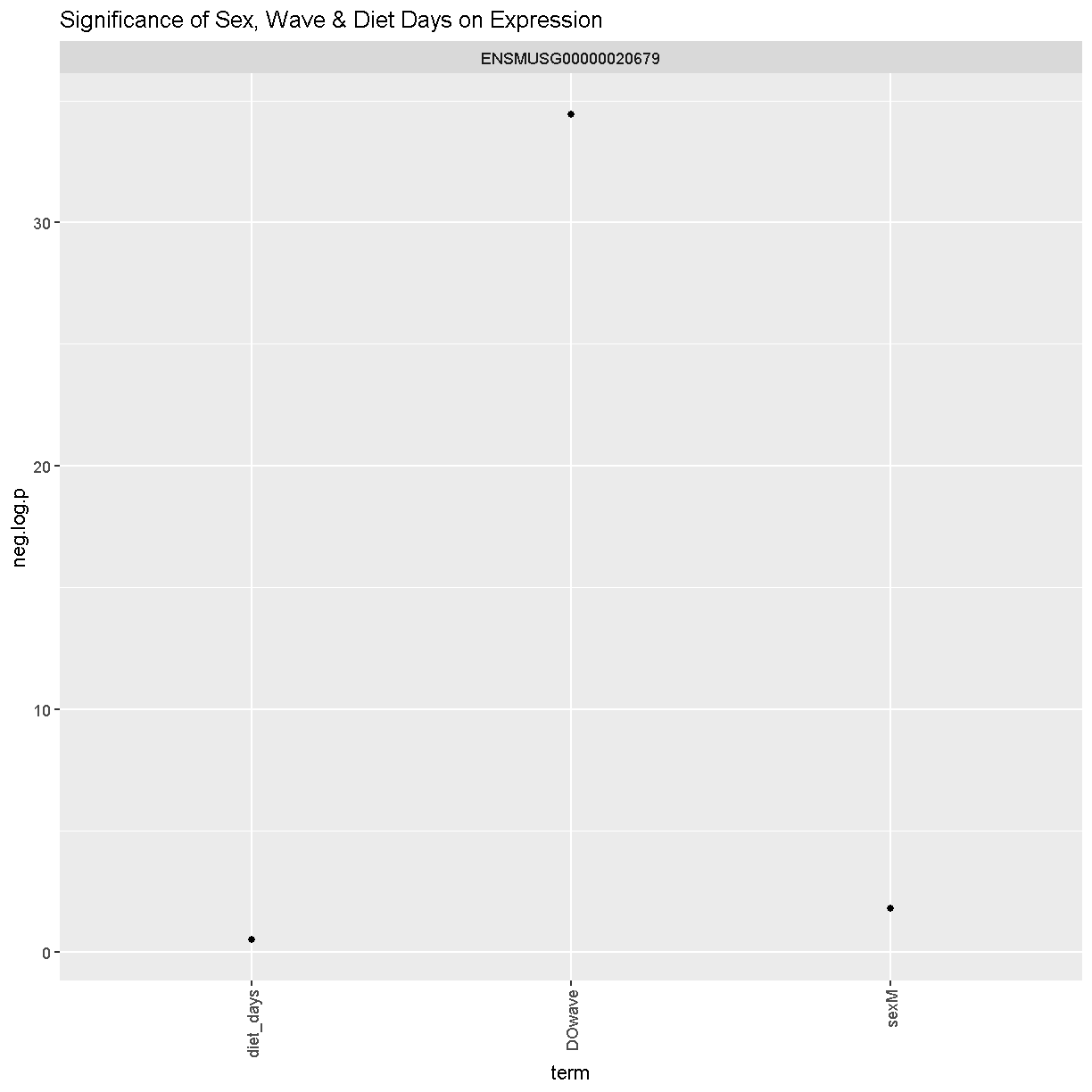
We can see that sex and DOwave are significant. Here DOwave is the group or batch number as not all mice were submitted for genotyping at the same time. Because of this, we now have to correct for it. Considering the paper included the covariate, diet_days, we will include that as well.
# convert sex and DO wave (batch) to factors
pheno_clin$sex = factor(pheno_clin$sex)
pheno_clin$DOwave = factor(pheno_clin$DOwave)
pheno_clin$diet_days = factor(pheno_clin$DOwave)
covar = model.matrix(~sex + DOwave + diet_days, data = pheno_clin)[,-1]
Performing a genome scan
[Permutations]
First, we need to work out the signifcance level. Let’s find the signifance level for 0.1, 0.05 and 0.01.
operm <- scan1perm(genoprobs = probs,
pheno = norm[,"ENSMUSG00000020679", drop = FALSE],
addcovar=covar,
n_perm=1000)
Note DO NOT RUN THIS (it will take too long). Instead, I have run it earlier and will load it in here. We will also perform a summary to find the summary level for 0.1, 0.05 and 0.01
load("../data/operm_ENSMUSG00000020679_1000.Rdata")
summary(operm,alpha=c(0.1, 0.05, 0.01))
LOD thresholds (1000 permutations)
ENSMUSG00000020679
0.1 7.29
0.05 7.73
0.01 8.30
Genome Scan
qtl = scan1(genoprobs = probs,
pheno = norm[,"ENSMUSG00000020679", drop = FALSE],
kinship = K,
addcovar = covar)
Next, we plot the genome scan.
plot_scan1(x = qtl,
map = map,
lodcolumn = "ENSMUSG00000020679",
main = colnames(qtl))
add_threshold(map, summary(operm, alpha=0.1), col = 'purple')
add_threshold(map, summary(operm, alpha=0.05), col = 'red')
add_threshold(map, summary(operm, alpha=0.01), col = 'blue')
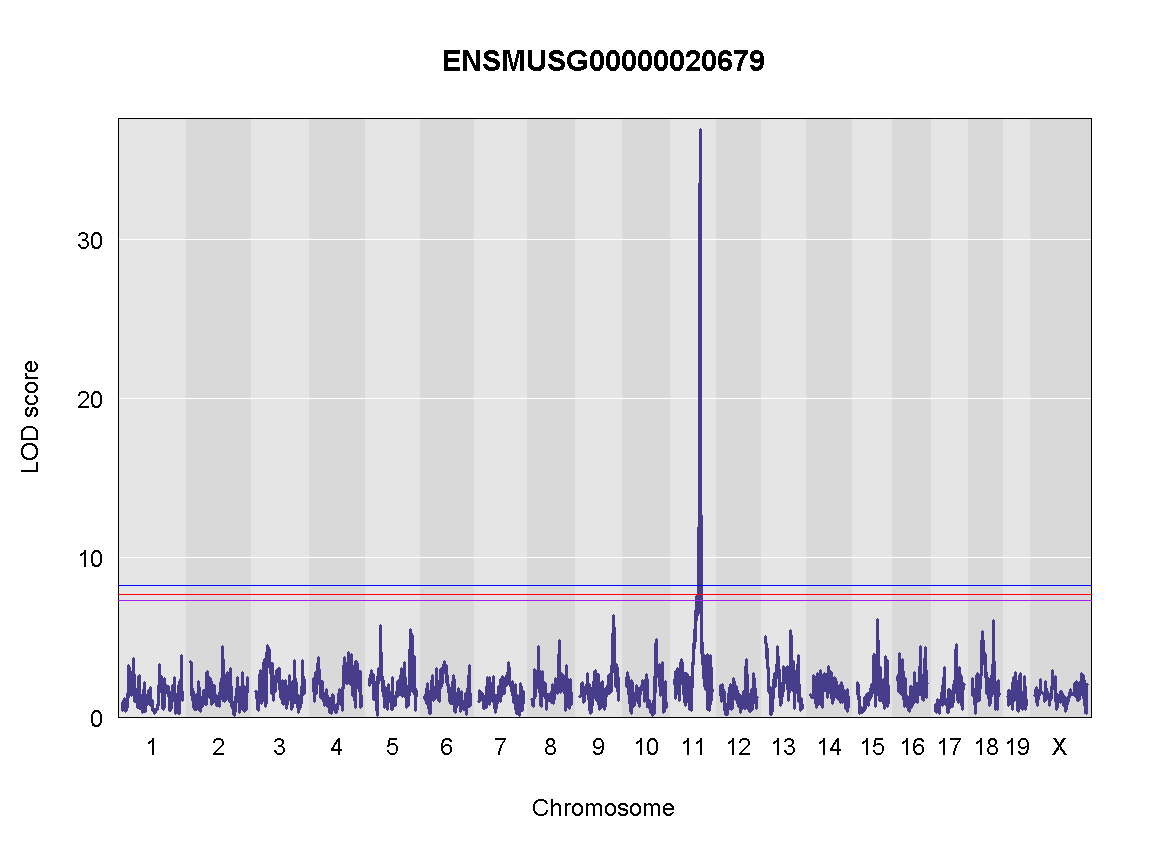
Finding LOD peaks
Let’s find LOD peaks
lod_threshold = summary(operm, alpha=0.01)
peaks = find_peaks(scan1_output = qtl,
map = map,
threshold = lod_threshold,
peakdrop = 4, prob = 0.95)
kable(peaks %>%
dplyr::select(-lodindex) %>%
arrange(chr, pos), caption = "Phenotype QTL Peaks with LOD >= 6")
Table: Phenotype QTL Peaks with LOD >= 6
| lodcolumn | chr | pos | lod | ci_lo | ci_hi |
|---|---|---|---|---|---|
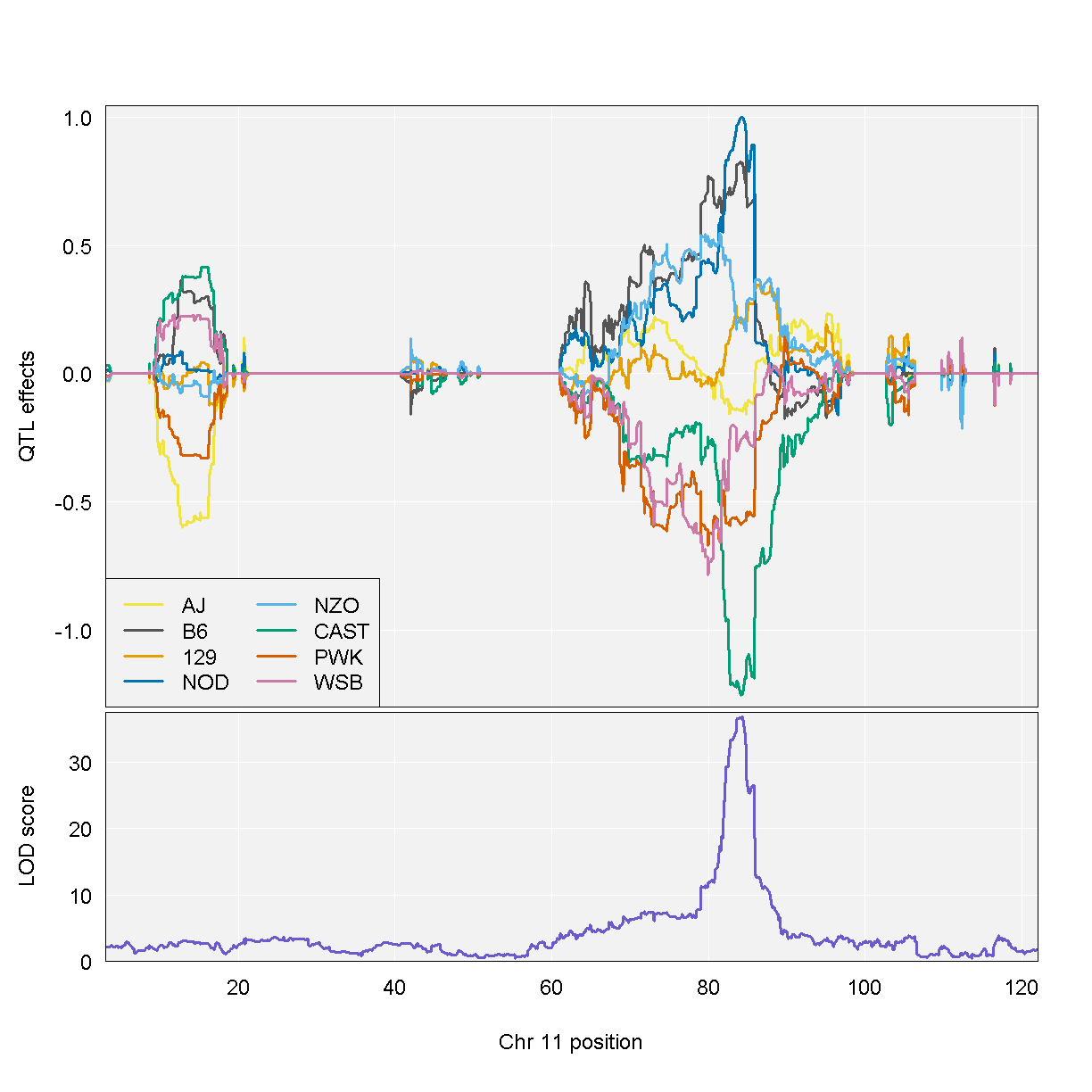
| ENSMUSG00000020679 | 11 | 84.40138 | 36.87894 | 83.64714 | 84.40138 |
QTL effects
blup <- scan1blup(genoprobs=probs[,peaks$chr[1]],
norm[,peaks$lodcolumn[1], drop=FALSE])
plot_coefCC(blup,
map=map,
columns=1:8,
bgcolor="gray95",
legend="bottomleft",
scan1_output = qtl )
Challenge
Now choose another gene expression trait in
normdata object and perform the same steps.
1). Check the distribution of the raw counts and normalised counts 2). Are there any sex, batch, diet effects?
3). Run a genome scan with the genotype probabilities and kinship provided.
4). Plot the genome scan for this gene.
5). Find the peaks above LOD score of 6.Solution
Replace
<ensembl id>with your choice of gene expression trait#1). hist(pheno_clin$<ensembl id>) pheno_clin$<ensembl id>_log <- log(pheno_clin$<ensembl id>) hist(pheno_clin$<ensembl id>_log) #2). tmp = pheno_clin %>% dplyr::select(mouse, sex, DOwave, diet_days, <ensembl id>) %>% gather(expression, value, -mouse, -sex, -DOwave, -diet_days) %>% group_by(expression) %>% nest() mod_fxn = function(df) { lm(value ~ sex + DOwave + diet_days, data = df) } tmp = tmp %>% mutate(model = map(data, mod_fxn)) %>% mutate(summ = map(model, tidy)) %>% unnest(summ) tmp tmp %>% filter(term != "(Intercept)") %>% mutate(neg.log.p = -log10(p.value)) %>% ggplot(aes(term, neg.log.p)) + geom_point() + facet_wrap(~expression) + labs(title = "Significance of Sex, Wave & Diet Days on Phenotypes") + theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5)) + rm(tmp) #3). qtl = scan1(genoprobs = probs, pheno = pheno_clin[,"<ensembl id>", drop = FALSE], kinship = K, addcovar = covar) #4). plot_scan1(x = qtl, map = map, lodcolumn = "<ensembl id>") abline(h = 6, col = 2, lwd = 2) #5). peaks = find_peaks(scan1_output = qtl, map = map, threshold = lod_threshold, peakdrop = 4, prob = 0.95)
Key Points
To run a QTL analysis for expression data