Creating A Transcriptome Map
Overview
Teaching: 30 min
Exercises: 30 minQuestions
How do I create and interpret a transcriptome map?
Objectives
Describe a transcriptome map.
Interpret a transcriptome map.
Load Libraries
library(tidyverse)
library(qtl2)
library(qtl2convert)
library(RColorBrewer)
library(qtl2ggplot)
source("../code/gg_transcriptome_map.R")
Load Data
Load in the LOD peaks over 6 from previous lesson.
# REad in the LOD peaks from the previous lesson.
lod_summary <- read.csv("../results/gene.norm_qtl_peaks_cis.trans.csv")
In order to use the ggtmap function, we need to provide specific column names. These are documented in the “gg_transcriptome_map.R” file in the code directory of this workshop. The required column names are:
- data: data.frame (or tibble) with the following columns:
- ensembl: (required) character string containing the Ensembl gene ID.
- qtl_chr: (required) character string containing QTL chromsome.
- qtl_pos: (required) floating point number containing the QTL position in Mb.
- qtl_lod: (optional) floating point number containing the LOD score.
- gene_chr: (optional) character string containing transcript chromosome.
- gene_start: (optional) character string containing transcript start postion in Mb.
- gene_end: (optional) character string containing transcript end position in Mb.
# Get gene positions.
ensembl <- get_ensembl_genes()
df <- data.frame(ensembl = ensembl$gene_id,
gene_chr = seqnames(ensembl),
gene_start = start(ensembl) * 1e-6,
gene_end = end(ensembl) * 1e-6,
stringsAsFactors = F)
lod_summary <- lod_summary %>%
rename(lodcolumn = "ensembl",
chr = "qtl_chr",
pos = "qtl_pos",
lod = "qtl_lod") %>%
left_join(df, by = "ensembl") %>%
mutate(marker.id = str_c(qtl_chr, qtl_pos * 1e6, sep = "_"),
gene_chr = factor(gene_chr, levels = c(1:19, "X")),
qtl_chr = factor(qtl_chr, levels = c(1:19, "X")))
rm(df)
Some of the genes will have a QTL in the same location as the gene and others will have a QTL on a chromosome where the gene is not located.
Challenge
a. What do we call eQTL that are co-colocated with the gene? b. What do we call eQTL that are located on a different chromosome than the gene?
Solution
a. A cis-eQTL is an eQTL that is co-colocated with the gene. b. A trans-eQTL is an eQTL that is located on a chromosome other than the gene that was mapped.
We can tabluate the number of cis- and trans-eQTL that we have and add this to our QTL summary table. A cis-eQTL occurs when the QTL peaks is directly over the gene position. But what if it is 2 Mb away? Or 10 Mb? It’s possible that a gene may have a trans eQTL on the same chromosome if the QTL is “far enough” from the gene. We have selected 4 Mb as a good rule of thumb.
lod_summary <- lod_summary %>%
mutate(cis = if_else(qtl_chr == gene_chr & abs(gene_start - qtl_pos) < 4, "cis", "trans"))
count(lod_summary, cis)
cis n
1 cis 33
2 trans 55
Plot Transcriptome Map
ggtmap(data = lod_summary %>% filter(qtl_lod >= 7.18), cis.points = TRUE, cis.radius = 4)
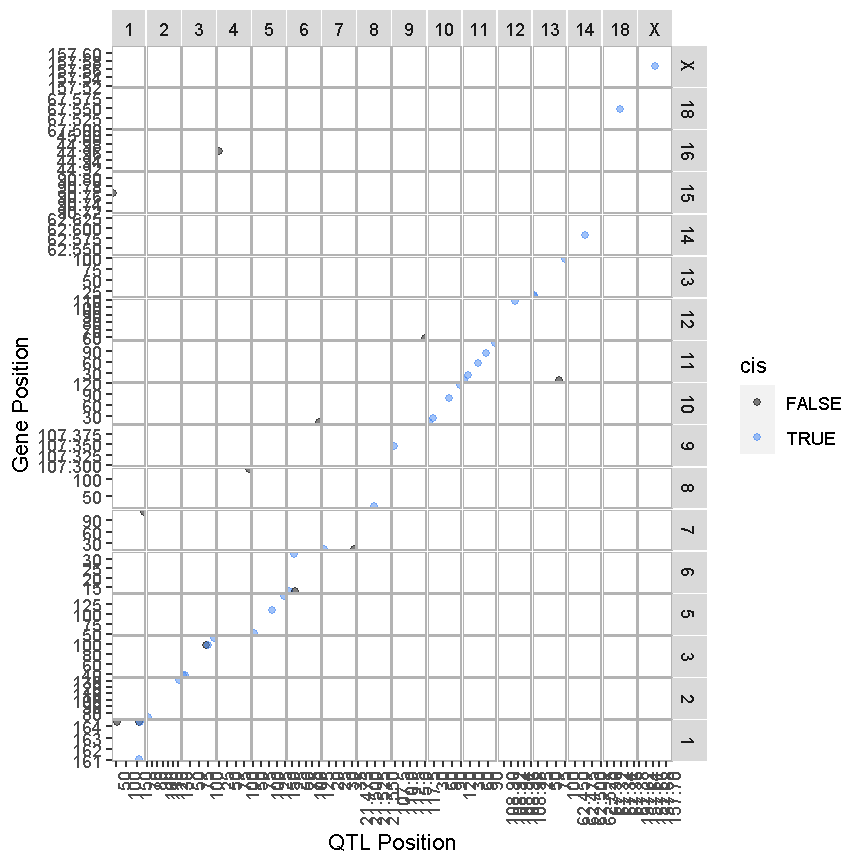
The plot above is called a “Transcriptome Map” because it shows the postions of the genes (or transcripts) and their corresponding QTL. The QTL position is shown on the X-axis and the gene position is shown on the Y-axis. The chromosomes are listed along the top and right of the plot. What type of QTL are the genes with QTL that are located along the diagonal?
Key Points
Transcriptome maps aid in understanding gene expression regulation.